Blog
Fecha publicación: 20-05-2012
Autor: Maria Jose Gil Moreno
"La Dra. María José Gil ha sido invitada a la presentación de un resumen de la patología neurológica por la que ha recibido el premio al mejor caso en demencias dentro del concurso de neurocasos"
La parálisis supranuclear progresiva es una enfermedad degenerativa del sistema nervioso central de curso progresivo descrita en 1964. Se caracteriza por alteraciones posturales, con caídas frecuentes, parkinsonismo rígido-acinético de predominio axial, alteración oculomotora y parálisis pseudobulbar.
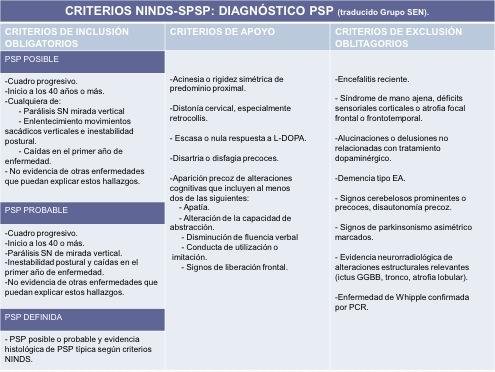
Los criterios diagnósticos son sensibles pero poco específicos. Los más utilizados son los propuestos por el National Institute of Neurological Disorders and Stroke y Society for Progressive Supranuclear Palsy (NINDS-SPSP).
Desde el punto de vista anatomo-patológico los hallazgos característicos de la PSP son:
- Estudio macroscópico: atrofia mesencéfalo, pedúnculo cerebeloso superior, protuberancia, globo pálido y núcleos subtalámicos. Despigmentación sustancia nigra, locus coeruleus y núcleo dentado del cerebelo.
- Estudio microscópico: inclusiones de proteína tau en astrocitos (astrocitos “en penacho”) y en oligodendrocitos (“cuerpos espirales”) localizados en ganglios de la base, diencéfalo, mesencéfalo y regiones perirrolándicas. Existen ovillos neurofibrilares con apariencia globosa.
Se han descrito formas atípicas en la Parálisis Supranuclear Progresiva, inicialmente descritas “formas frustradas” por Steele, Richardson y Olszewski. Lantos en 1994 describe tres tipos de PSP, según la distribución de patología neurofibrilar. En 2005, Williams describe dos variantes, la forma parkinsoniana que responde a L-DOPA (PSP-P) y la forma de aquinesia pura y fallo de la marcha (PSP-PAGF). Posteriormente Williams describe dos tipos de variantes: corticales y de tronco cerebral, según la distribución de patología tau.
La variante cortical puede presentarse en forma de demencia frontotemporal, degeneración corticobasal, afasia progresiva no fluente y esclerosis lateral primaria y se caracteriza por la presencia de patología tau en áreas corticales. En la variante de tronco cerebral se engloba el parkinsonismo con respuesta a L-Dopa y la forma aquinesia pura y fallo de la marcha, con patología tau en globo pálido, diencéfalo y tronco cerebral.
Existe por tanto una forma de Parálisis Supranuclear Progresiva que puede presentarse con síntomas similares a una Demencia Frontotemporal y que presenta un sustrato patológico de acúmulo de proteína tau en áreas corticales y otras áreas típicas de PSP.
Diversos estudios de presentación frontal en PSP afirman que la variante conductual suele ser la forma más frecuente y que el inicio del diagnóstico suele ser a edad más temprana y con síntomas motores de inicio.
La parálisis supranuclear progresiva es la taupatía 4R más frecuente. Existe una gran heterogenicidad clínica dentro de las taupatías; con relativa frecuencia existen síndromes “solapados”, con características clínicas y patológicas similares, como el caso que presentamos. Es importante el uso de criterios clínicos que deben ser utilizados como guía para el diagnóstico de estas entidades y confirmar con el examen patológico la sospecha clínica.
BIBLIOGRAFIA
1. Steele J.C.; Richardson J.C.; Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Archives of Neurology 1964; 10: 333-359.
2. Carnero Pardo C. Parálisis supranuclear progresiva. En: Guía oficial para la práctica clínica en demencias: conceptos, criterios y recomendaciones 2009. Grupo de Estudio de Neurología de la Conducta y Demencias. Sociedad Española de Neurología. Barcelona. Editorial: Prous Science, S.A.U. 2009.p.143-154.
3. Litvan I.; Agid Y.; Calnde D. et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology 1996; 47: 1-9.
4. Cains N.J.; Bigio E.H.; Mackencie I.R. et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathology 2007 July; 114 (1): 5-22.
5. Lantos PL. The neuropathology of progressive supranuclear palsy. J Neural Transm 1994; 42:137-152.
6. Williams D.R.; de Silva R.; Paviour D.C, et al. Characteristics of two distinct clinical phenotypes in pathologically proven progressive supranuclear palsy: Richardson´s syndrome and PSP-parkinsonism. Brain 2005; 128:1247-1258.
7. Williams D.R.; Lees A. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurology 2009; 8: 270-279.
8. Dickson D.W.; Ahmed Z.; Algom A.A.; Tsuboi Y.; Josephs K.A. Neuropathology of variants of progressive supranuclear palsy. Current Opinion Neurology 2010; 23: 304-400.
9. Kaat D.L; Boon A.J.; Kamphorst W.; Duivenvoorden H.J.; van Swieten J.C. Frontal presentation in progressive supranuclear palsy. Neurology 2007; 69: 723-729.
10. Bibio E.H; Brown D.F.; White III C. Progressive supranuclear palsy with Dementia: Cortical Pahology. Journal of Neuropathology and Experimental Neurology 1999; 58: 359-364.
- Inicie sesión para enviar comentarios
- 22083 lecturas
- Todos los artículos de Maria Jose Gil Moreno


Comentarios
¿Hacia un origen común de las enfermedades neurodegenerativas?
Éste es un buen ejemplo de cómo distintas enfermedades neurodegenerativas pueden iniciarse de una forma similar aunque la histología corresponda a una taupatía (como el caso de la PSP) o bien tener otro tipo de alteración histológica. El "overlap" entre la parálisis supranuclear progresiva y la degeneración cortico-basal es bastante frecuente y se conoce como síndrome PSP-DCB. No obstante el inicio "conductual", como el caso presentado por la Dra. Gil, tampoco es infrecuente y establece la conexión con las degeneraciones fronto-temporales (obviamente con aquellas con histología de acumulo de proteína tau).
El espectro sintomático de las enfermedades neurodegenerativas con síntomas motores, cognitivos y conductuales sólo nos proporciona algunas claves sobre la histología subyacente, pero poco más... en definitiva un "síndrome conductual" con disfunción ejecutiva puede esconder (casi) cualquier tipo de histología, incluso placas amiloides y ovillos neurofibrilares típicos de la enfermedad de Alzheimer...
Quizás, todo esto nos haga reflexionar sobre la posibilidad de que las distintas enfermedades neurodegenerativas escondan un mecanismo fisiopatológico común; o bien, que la clínica no sea más que la expresión de la topografía de unas lesiones que pueden distribuirse con cierto grado de aleatoriedad.
En fin, esperaremos los nuevos descubrimientos de la neurociencia básica con impaciencia.